




| Original Article | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sudan J Paed. 2023; 23(2): 153-162 SUDANESE JOURNAL OF PAEDIATRICS 2023; Vol 23, Issue No. 2 ORIGINAL ARTICLE Electrophysiology of ataxia-telangiectasia-like disorder 1Salah A. Elmalik (1)(1) Department of Physiology, College of Medicine, King Saud University, Riyadh, Saudi Arabia Correspondence to: Salah A Elmalik Department of Physiology, College of Medicine, King Saud University, Riyadh, Saudi Arabia. Email: salah.elmalik2 [at] gmail.com Received: 20 December 2020 | Accepted: 20 December 2023 How to cite this article: Elmalik SA. Electrophysiology of ataxia-telangiectasia-like disorder 1. Sudan J Paediatr. 2023;23(2):153–162. https://doi.org/10.24911/SJP.106-1703054783 © 2023 SUDANESE JOURNAL OF PAEDIATRICS
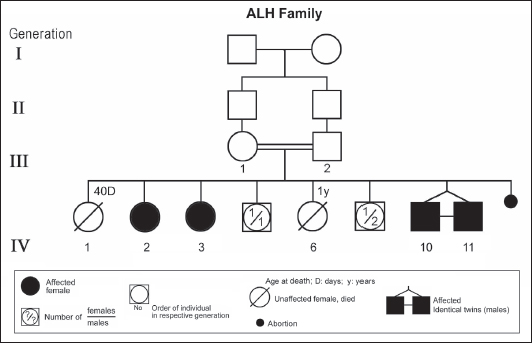
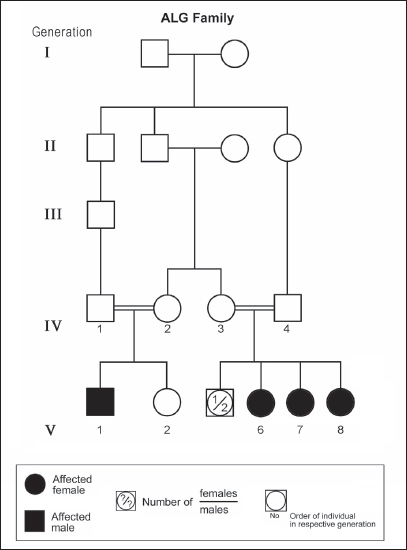
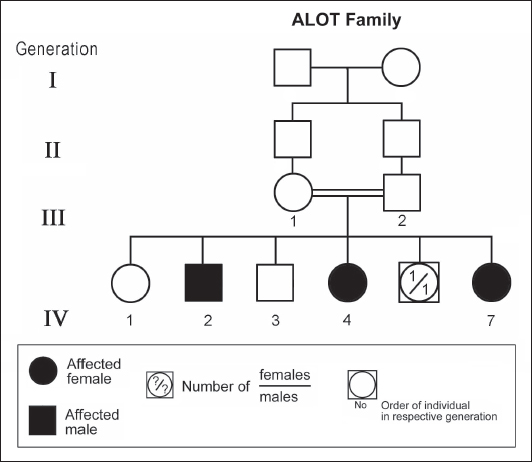
ABSTRACTAtaxia-telangiectasia-like disorder-1 (ATLD1, OMIM # 604391) is a very rare clinical condition, characterized by slowly progressive ataxia with onset in childhood, associated with oculomotor apraxia and dysarthria. Laboratory findings reveal increased susceptibility to radiation, with a defect in DNA repair. Patients with ATLD1 show no telangiectasia, have no immunodeficiency, and also have preserved cognition. Reflexes might be initially brisk and later becomes reduced associated with axonal sensorimotor neuropathy. Brain magnetic resonance imaging (MRI) detects cerebellar atrophy. The condition is caused by mutations in the meiotic recombination 11 (MRE11A) gene. The present study reports on the neurophysiologic finding in eight Saudi patients, belonging to three Saudi families, who have genetically confirmed ATLD1. All investigated patients had cerebellar atrophy on brain MRI (5/5). Electrophysiologic studies showed normal motor conduction velocity (MCV) of the median (8/8) and tibial (2/2) nerves, while 5/6 (83%) had normal peroneal nerve MCV. The distal motor latency (DML) for median, tibial, and peroneal nerves was within the normal range in all examined patients. The amplitude of compound muscle action potential (CMAP) of median and tibial nerves was also normal, while that of the peroneal nerve was normal in 3/6 (50%). Two of seven (29%) patients had reduced amplitude of median nerve sensory nerve action potential (SNAP) while 3/8 (38%) had a reduction in the amplitude of sural nerve SNAP. These findings favour an axonal type of neuropathy predominately affecting the sensory fibres (axonal sensorimotor neuropathy). The present study constitutes the largest cohort of ATLD1 patients worldwide who had electrophysiologic tests. KEYWORDSAtaxia-telangiectasia-like disorder-1 (ATLD1, OMIM # 604391); Oculomotor apraxia and dysarthria; Paediatrics. INTRODUCTIONAtaxia-telangiectasia-like disorder-1 (ATLD1, OMIM # 604391) is a very rare clinical condition that is characterized by slowly progressive ataxia with onset between 1 and 7 years of age, associated with oculomotor apraxia and dysarthria [1,2]. Laboratory findings show increased susceptibility to radiation, with a defect in DNA repair. Patients with ATLD1 show no telangiectasias, have no immunodeficiency, and serum alpha fetoprotein (AFP) concentrations are normal [3]. They also have preserved cognition [1,4]. Reflexes might be initially brisk and later becomes reduced. At advanced stages of the disease, tongue and facial dyskinesia, choreoathetosis, and dystonia, suggesting basal ganglia compromise, might be seen [4]. ATLD1 is progressive up to adolescence when it stabilizes [4]. There is no increased risk for infections or neoplasia, as is seen in ataxia-telangiectasia (A-T), but occasionally microcephaly is present [1], and pulmonary cancer was reported in two Japanese siblings [5]. Brain magnetic resonance imaging (MRI) detects cerebellar atrophy, and laboratory tests are non-informative [4]. Radiosensitivity test is usually present but in a lesser degree than in A-T [3]. ATLD1 is caused by mutations in the meiotic recombination 11 (MRE11) gene [6]. The largest subset of ATLD patients (15/25) was identified from Saudi Arabia [7]. Fernet et al. [1] described 10 patients, whereas Bohlega et al. [8]. reported another five. The remaining 10 patients included four in the UK (2 of them of Pakistani origin), two in Italy, and four in Japan [6,9–11]. Genetic studies showed that all Saudi patients had the same G to C transversion at position 630 (c.630G > C) located in exon 7, which results in the missense change of Trp to Cys (W210C). A study designed to assess the allelic frequency of this specific mutation in the Saudi population [12], found heterozygous genotype G/C of 0.4% and concluded that MRE11 should be explored in all cases of AT-like phenotype in Saudi Arabia. This resulted in the diagnosis later of the five patients reported by Bohlega et al. [8]. Chaki et al. [13] reported two sibs, who were born with ataxia and cerebellar vermis hypoplasia of consanguineous Pakistani parents. One of the patients had dysarthria and myoclonus. Miyamoto et al. [14] reported a 52-year-old woman who presented with progressive myoclonus of subcortical origin starting at age 9 years. She also presented with mild limb and truncal ataxia. Raslan et al. [15], reported two cases of ATLD: the first one with ataxia telangiectasia-like disorder type 1 related to MRE11A gene, while the second one had ataxia telangiectasia-like disorder type 2 related to PCNA gene. The first case is an 8-year girl born to non-consanguineous parents who presented with slow progression of gait ataxia which started at 2 years. Electromyography (EMG) was done for the patient and showed axonal neuropathy. Whole exome sequencing (WES) identified two heterozygous variants in the MRE11A gene NM_005590.4 (MRE11):c.1876_1895dup (p.Lys633fs) and NM_005590.4(MRE11):c.1516G > T (p.Glu506Ter). Hereditary ataxias are frequently associated with peripheral neuropathy. Axonal sensorimotor neuropathy is found in ataxia with oculomotor apraxia type 1, ataxia with oculomotor apraxia type 2, ataxia telangiectasia, cerebrotendinous xanthomatosis, Refsum’s disease, and Spastic ataxia of Charlevoix Saguenay. Pure sensory neuropathy is a feature of Friedreich ataxia, AVED, betalipoproteinaemia, SANDO, while no neuropathy is seen in ARCA1, ARCA2, SCA6, SCA8, and DPRLA [16]. Chronic denervation on EMG is a frequent finding in juvenile and adult onset hexosainidase A deficiency [17]. In mitochondrial disorders, the patients may have myopathic EMG findings with sensory neuropathy [18]. External sphincter EMG and autonomic function tests may be useful in the diagnosis of multiple systems atrophy [19]. and electroencephalogram (EEG) findings are useful in the diagnosis of sporadic and familial prion disease [20]. The present study reports on the neurophysiologic finding in eight Saudi patients, belonging to three Saudi families, who have genetically confirmed ATLD1. To the best of our knowledge, This study constitutes the largest cohort of ATLD1 patients worldwide who had electrophysiologic tests. MATERIALS AND METHODSSubjectsThis study included eight patients; all were Saudi. The age of onset was below 13 years in all patients. All cases were referred from Pediatric Neurology Clinics of King Khalid University Hospital (KKUH), College of Medicine, Riyadh. The patients were evaluated using a comprehensive protocol which included clinical examination and relevant laboratory data. Salient information about each child was retrieved. This included gender, history of the disease and other associated medical disorders, and physical signs. Consanguinity of parents and any family history of similar conditions, as well as laboratory data were filled for each patient. Pedigrees of affected families were drawn using the standard pedigree symbol. MRI was performed for at least one patient per family. It was also done for cases of inherited peripheral neuropathy with associated cognitive involvement. These were reviewed by both neuroradiologists and a paediatric neurologist. Genetic analysisPeripheral blood samples were collected from each patient, their living parents, and most of their unaffected siblings. Informed consent was obtained from all participants. Genomic DNA was extracted from peripheral blood leukocytes using Qiagen DNA isolation kits. DNA was sent for genetic analysis to the Institute of Genetics and Molecular and Cellular Biology, University of Strasbourg, and College de France, Illkirch; and the Laboratory of Neurogenetics, Department of Genetics, Cytogenetics and Embryology, la Pitié-Salpêtrière Hospital, Paris, France. Genome-wide linkage studies, microsatellite marker analysis, and mutational analysis were performed as described [1,21–25]. Neurophysiology testsThe neurophysiology tests were done at the Neurophysiology Laboratory, Division of Clinical Neurophysiology, Department of Neuroscience, Armed Forces Hospital, Riyadh. All children had at least one electrodiagnostic evaluation performed at the time of referral. Nerve conduction studies (NCS) were performed using the Nicolet EMG-NCS machine. The records were obtained following a conventional protocol [26]. Surface electrodes and stimulators were used for NCS. Amplitudes of compound muscle action potential (CMAPs), distal latency (DL), and conduction velocity were recorded whenever possible from median, ulnar, peroneal, and tibial nerves using conventional methods. The CMAP amplitudes for each motor nerve were measured from the peak of the negative wave to the baseline, and for sensory nerves from peak-to-peak. Temperature was controlled and was never less than 33°C. Median and ulnar CMAPs were recorded from the abductor pollicis brevis and the abductor digiti minimi, respectively, with stimulation at the wrist and elbow. The peroneal CMAP was recorded from the extensor digitorum brevis with stimulation at the ankle and knee. The tibial CMAPs were recorded from abductor hallucis with stimulation at the ankle and knee. DML, motor nerve conduction velocity potential (MNCV) and distal CMAPs amplitude were measured. In selective cases CMAPs and DML from the diaphragm muscle were recorded by using phrenic nerve stimulation in the neck [27]. Facial nerve CMAPs and DML were obtained by stimulation below and in front of the pinna of the ear and recorded from the nasalis muscle. Median sensory nerve action potentials (SNAPs) were obtained orthodromically at the wrist, following stimulation of the second digit. The sural SNAP was recorded antidromically at the ankle following stimulation of the calf. SNAP amplitudes and DL were measured. The results were recorded and evaluated using normal values for our lab [28–30]. Statistical analysisStat Pac Gold Statistical Analysis Package was used for data management. Mean, median, and standard deviation were calculated. Kruskal–Wallis test was used for comparing variables when the sample size was small. A probability value of <0.05 was considered to be significant. RESULTSEight patients with ATLD1, belonging to three Saudi families, were clinically evaluated, investigated, and also had neurophysiologic testing. The first Saudi family (ALH Family, Figure 1) had four affected individuals consisting of two females and two male twins. The parents, who were first-degree cousins, lost two daughters who died at 40 days and one year of age, respectively, from unknown causes. The second family (ALG Family, Figure 2) had three affected females, two of whom (V.6 and V.8) were available for the study. There was also history of a maternal cousin (V.1, Figure 2) who was similarly affected. The third Saudi family (ALOT family, Figure 3) had three affected patients, two of whom (IV.2 and IV.4) were available for clinical and electrophysiologic testing.
Figure 1. Pedigree of ALH family. Standard pedigree symbols are used.
Figure 2. Pedigree of ALG family. Standard pedigree symbols are used.
Figure 3. Pedigree of ALOT family. Standard pedigree symbols are used. The eight patients who were studied consisted of five females and three males. Their clinical features and investigations are summarized in Table 1. The mean age at assessment was 21.3 years (median=19.5 years, range=9–39 years). The onset of ataxia was early in life at a mean age of 2.1 years (median=1.3 years; range=1–7 years). The disease was relentlessly progressive and two patients in one family (IV.2 and IV.3, ALH Family, Figure 1) lost ambulation, each at the age of 13 years. On examination, all affected individuals had features of ataxia associated with oculomotor apraxia, but no telangiectasia. Two parents belonging to one family (V.6 and V.8, ALG Family, Figure 2) were microcephalic and both had low IQs (72 and 78, respectively). No patient examined had recurrent pulmonary infections, lymphoid or other tumours, skin abnormalities, muscle weakness, or lost sensation for crude touch. The four oldest patients in the group, all of whom belonged to one family (ALH Family, Figure 1), had signs of basal ganglia involvement manifesting as facial dyskinesia (abnormal and involuntary facial movements), and masked face. Deep tendon reflexes were brisk at 9 and 13 years, respectively, in the two patients belonging to one family (V.6 and V.8, ALG Family, Figure 2). They were reduced in individuals of the other two families, who were relatively older (age range=13–39 years). All investigated patients showed cerebellar atrophy on brain MRI (5/5) and had normal levels of alpha-fetoprotein (6/6), carcinoembryonic antigen (5/5), and immunoglobulins (6/6). Also, all those investigated had normal levels of serum albumin (7/7) and cholesterol (5/5), including the patients of the ALH Family (Figure 1) who have had clinical symptoms of the disease for >20 years. In all affected individuals of the three families, molecular genetic testing revealed homozygous missense mutation (630G®C, W210C) of the MRE 11 gene. Table 1. Clinical, biochemical, and neuroimaging features of three families with ataxia telangiectasia-like disorder1.
aSkull circumference; +=present; -=absent. F, female; IQ, intelligence quotient; M, male; MRI, magnetic resonance imaging; N, normal; NA, not ascertained; SD, standard deviation. ElectrophysiologyNCS was tested in all patients. MCV, DML, and amplitudes of CMAPs were recorded from the median nerve in eight patients, peroneal in six patients, and tibial nerve in two patients, Recordings of SNAPs, whenever possible, were done from the median nerve in seven patients and sural nerve in eight patients. The result of electrophysiology is given in Table 2. MCV of the median nerve was normal in all patients (mean: 60.87 ± 9.01 m/s). DML of the median nerve was normal in all of them (mean: 3.02 ± 0.37 ms), while amplitude CMAPs of the median was also normal in all patients (mean: 6.25 ± 1.78 mV). Peroneal nerve MCV was normal in five out of six patients (83%) [mean: 53.48 ± 19.04 m/s], DML of peroneal was within normal range in all six tested patients (mean: 4.26 ± 0.48 ms), while amplitude of CMAPs of peroneal was normal in 3/6 (50%) and reduced in the rest of the patients (mean: 2.18 ± 1.61 mV). Tibial nerve was tested in two patients (P4 and P5) and was found to have normal MCV in both patients (42 and 45 m/s), with normal DML (4.2 and 3.8 ms) and normal amplitude of CMAPs (4.1 and 11.3 mV). The amplitude of SNAPs of the median nerve was normal in 2/7 patients (29%) while it was either not recordable or reduced in the rest of the tested patients (mean: 14.91 ± 12.25 μV), while the amplitude of SNAPs of the sural nerve was normal in 3/8 patients (38%), and in 5 patients it was either not recordable or reduced (mean: 22.4 ± 16.65 μV). DISCUSSIONEight patients with ataxia telangiectasia-like disorder1 (ATLD1), belonging to three Saudi families, were clinically evaluated, investigated, and also had electrophysiologic testing. On examination, they showed features of ataxia associated with oculomotor apraxia, but no telangiectasia [1,2]. All investigated patients revealed cerebellar atrophy on brain MRI [5/5] and had normal levels of alpha-fetoprotein (6/6) and immunoglobulins (6/6). Clinically, ATLD1 is very similar to ataxia telangiectasia. However, patients with ATLD1 show no telangiectasias, have no immunodeficiency, and serum alpha-fetoprotein concentrations are normal [3]. Electrophysiologic studies showed normal MCV of the median (8/8) and tibial (2/2) nerves in all patients, while 5/6 (83%) had normal peroneal nerve MCV. The DML for median, tibial, and peroneal nerves was within the normal range in all examined patients. The amplitude of CMAP of median and tibial nerves was also normal, while that of the peroneal nerve was normal in 3/6 (50%). Two of seven (29%) patients had reduced amplitude of median nerve SNAP while 3/8 (38%) had a reduction in the amplitude of sural nerve SNAP. These findings favor an axonal type of neuropathy predominately affecting the sensory fibers (axonal sensorimotor neuropathy) similar to what has been reported in the literature [8]. Bohlega et al. [8] described five patients with ATLD who belonged to two families. One of these (Family B) had normal tendon reflexes and normal NCSs, whereas vibration senses were decreased distally and tendon reflexes were absent in the other family (Family D). The largest cohort of ATLD1 patients worldwide was identified in Saudi Arabia and included 10 patients [1], but no electrophysiological study was done for this group. Eight of these patients were followed up in the present study, and constitute the largest cohort of ATLD1 patients worldwide who had electrophysiologic tests. Genetic studies showed that all Saudi patients with ATLD1 had the same missense mutation (a G-to-C change at nucleotide 630) in the MRE11 gene. A heterozygous carriers’ frequency of 0.4% was recorded in the Saudi population by Alsbeih et al. [7] who concluded that MRE11 should be explored in all cases of AT-like phenotype in Saudi Arabia. This resulted in the diagnosis later of the 5 patients reported by Bohlega et al. [8]. Table 2. Electrophysiology of three families with ataxia telangiectasia-like disorder1.
-=not done. CMAPs, Compound muscle action potentials; MNCV, Motor nerve conduction velocity potential; NR, Not recordable; y, Years. CONCLUSIONElectrophysiologic study findings showed an axonal type of neuropathy that mainly affects sensory fibers (axonal sensorimotor neuropathy). These findings are consistent with what has been reported in the literature. In the present study, electrophysiologic investigations were done for eight patients with ataxia telangiectasia-like disease1 (ATLD1); and this constitutes the largest cohort of ATLD1 patients worldwide who had electrophysiologic tests. Conflict of interestThe author declares no conflicts of interest. FundingNone. Ethical approvalSigned informed consent for participation and publication of medical details was obtained from the patients and from the parents of children. Ethics committee approval was obtained from the College of Medicine Research Board, King Saud University, Riyadh, Saudi Arabia. REFERENCES
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| How to Cite this Article |
| Pubmed Style Salah A Elmalik. Electrophysiology of ataxia-telangiectasia-like disorder 1. Sudan J Paed. 2023; 23(2): 153-162. doi:10.24911/SJP.106-1703054783 Web Style Salah A Elmalik. Electrophysiology of ataxia-telangiectasia-like disorder 1. https://sudanjp.com//?mno=182010 [Access: February 05, 2026]. doi:10.24911/SJP.106-1703054783 AMA (American Medical Association) Style Salah A Elmalik. Electrophysiology of ataxia-telangiectasia-like disorder 1. Sudan J Paed. 2023; 23(2): 153-162. doi:10.24911/SJP.106-1703054783 Vancouver/ICMJE Style Salah A Elmalik. Electrophysiology of ataxia-telangiectasia-like disorder 1. Sudan J Paed. (2023), [cited February 05, 2026]; 23(2): 153-162. doi:10.24911/SJP.106-1703054783 Harvard Style Salah A Elmalik (2023) Electrophysiology of ataxia-telangiectasia-like disorder 1. Sudan J Paed, 23 (2), 153-162. doi:10.24911/SJP.106-1703054783 Turabian Style Salah A Elmalik. 2023. Electrophysiology of ataxia-telangiectasia-like disorder 1. Sudanese Journal of Paediatrics, 23 (2), 153-162. doi:10.24911/SJP.106-1703054783 Chicago Style Salah A Elmalik. "Electrophysiology of ataxia-telangiectasia-like disorder 1." Sudanese Journal of Paediatrics 23 (2023), 153-162. doi:10.24911/SJP.106-1703054783 MLA (The Modern Language Association) Style Salah A Elmalik. "Electrophysiology of ataxia-telangiectasia-like disorder 1." Sudanese Journal of Paediatrics 23.2 (2023), 153-162. Print. doi:10.24911/SJP.106-1703054783 APA (American Psychological Association) Style Salah A Elmalik (2023) Electrophysiology of ataxia-telangiectasia-like disorder 1. Sudanese Journal of Paediatrics, 23 (2), 153-162. doi:10.24911/SJP.106-1703054783 |


Nagwa Salih, Ishag Eisa, Daresalam Ishag, Intisar Ibrahim, Sulafa Ali
Sudan J Paed. 2018; 18(1): 24-27
» Abstract » doi: 10.24911/SJP.2018.1.4
Siba Prosad Paul, Emily Natasha Kirkham, Katherine Amy Hawton, Paul Anthony Mannix
Sudan J Paed. 2018; 18(2): 5-14
» Abstract » doi: 10.24911/SJP.106-1519511375
Inaam Noureldyme Mohammed, Soad Abdalaziz Suliman, Maha A Elseed, Ahlam Abdalrhman Hamed, Mohamed Osman Babiker, Shaimaa Osman Taha
Sudan J Paed. 2018; 18(1): 48-56
» Abstract » doi: 10.24911/SJP.2018.1.7
Adnan Mahmmood Usmani; Sultan Ayoub Meo
Sudan J Paed. 2011; 11(1): 6-7
» Abstract
Mustafa Abdalla M. Salih, Mohammed Osman Swar
Sudan J Paed. 2018; 18(1): 2-5
» Abstract » doi: 10.24911/SJP.2018.1.1
Amir Babiker, Afnan Alawi, Mohsen Al Atawi, Ibrahim Al Alwan
Sudan J Paed. 2020; 20(1): 13-19
» Abstract » doi: 10.24911/SJP.106-1587138942
Bashir Abdrhman Bashir, Suhair Abdrahim Othman
Sudan J Paed. 2019; 19(2): 81-83
» Abstract » doi: 10.24911/SJP.106-1566075225
Anita Mehta, Arvind Kumar Rathi, Komal Prasad Kushwaha, Abhishek Singh
Sudan J Paed. 2018; 18(1): 39-47
» Abstract » doi: 10.24911/SJP.2018.1.6
Majid Alfadhel, Amir Babiker
Sudan J Paed. 2018; 18(1): 10-23
» Abstract » doi: 10.24911/SJP.2018.1.3
Amir Babiker, Mohammed Al Dubayee
Sudan J Paed. 2017; 17(2): 11-20
» Abstract » doi: 10.24911/SJP.2017.2.12
|
| |
About Sudanese Journal of PaediatricsSudanese Journal of Paediatrics is available free online at https://sudanjp.com/. Archives can also be found at https://sudanjp.org/. There ... Read more. For best results, please use Internet Explorer or Google Chrome. |
Contact InformationEditor in Chief: Prof Satti Abdulrahim Satti Mohamed, Professor of Pediatrics and Child Health. Pediatric Tropical & Infectious Disease Consultant, Faculty of Medicine, National University, Sudan. E-mail: sattiabd99@gmail.com Sudan Association of Paediatricians, Building No.71, Block No.65, Arkaweet, Khartoum, Sudan. Journal Email: editor.sudanjp@gmail.com Technical Support:anastasiia.pavlenco@sofiafields.comhttps://sofiafields.com/ Hosting Providor:contact@discoverpublish.comhttps://discoverpublish.com/ |